Beta talasemia adalah sebuah tantangan kesehatan yang serius, memengaruhi ribuan bayi setiap tahunnya di seluruh dunia. Memahami kondisi ini tidak hanya penting untuk mereka yang terdiagnosis, tetapi juga bagi masyarakat luas, mengingat dampak genetiknya yang dapat diwariskan.
Kasus Beta Talasemia: Global dan di Indonesia
Beta talasemia (beta talasemia) adalah salah satu jenis hemoglobinopati, yaitu kelainan darah yang diturunkan dan disebabkan oleh mutasi pada gen yang mempengaruhi produksi hemoglobin. Pada beta talasemia, mutasi ini terjadi pada gen beta globin, sehingga mengurangi jumlah hemoglobin fungsional yang dihasilkan. Akibatnya, sel darah merah mengalami kerusakan dan menyebabkan anemia.[1]
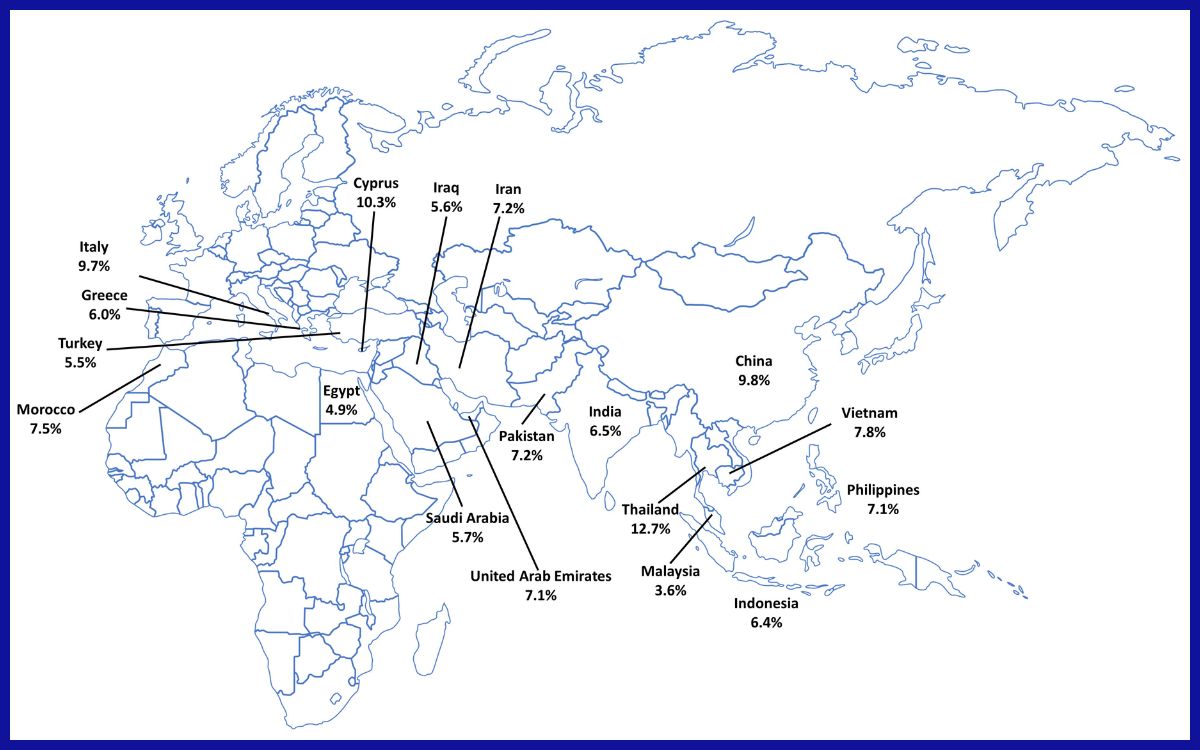
Tingkat pembawa sifat β-thalassemia di negara-negara endemik.[1]
Berdasarkan World Health Organization (WHO), secara global, sekitar 40.000 bayi lahir setiap tahun dengan talasemia, dan mayoritas kasusnya adalah beta talasemia. Sebagian besar anak-anak dengan talasemia lahir di negara-negara berpenghasilan rendah dan menengah, terutama di Asia Tenggara. Di Indonesia sendiri, sekitar 3,0–10,0% penduduk membawa beta talasemia, dengan perkiraan 2.500 bayi lahir dengan beta talasemia mayor setiap tahunnya.[1, 2]
Hubungan antara Beta Talasemia dan Genetika
Beta talasemia adalah kelainan darah yang diturunkan dan ditandai dengan adanya permasalahan pada produksi rantai beta-globin, yang merupakan komponen penting dari hemoglobin. Pemahaman tentang pola pewarisannya penting untuk mencegah penyakit kelainan tersebut.[3]
Beta talasemia terjadi akibat mutasi pada gen β-globin (HBB) yang terletak di kromosom 11. Mutasi ini dapat menyebabkan berkurangnya (β+) atau bahkan tidak adanya produksi rantai β-globin (β0). Gen HBB sendiri bertugas memberikan instruksi untuk membentuk β-globin, sebuah protein yang menjadi komponen penting dari hemoglobin, yaitu protein yang berada dalam sel darah merah. Hemoglobin memiliki peran penting dalam mengangkut oksigen ke seluruh tubuh.[3]
Tingkat keparahan beta talasemia sangat dipengaruhi oleh jenis mutasi yang terjadi. Hingga saat ini, telah diidentifikasi lebih dari 200 variasi genetik pada gen HBB. Kebanyakan kasus beta talasemia disebabkan oleh mutasi titik, yaitu penggantian atau penyisipan satu nukleotida. Namun, dalam kasus yang lebih jarang, beta talasemia juga bisa dipicu oleh mutasi delesi, yang menyebabkan hilangnya sebagian materi genetik.[3]
Tiga kondisi klinis pada beta talasemia yang diketahui berdasarkan tingkat keparahannya adalah: beta talasemia minor, talasemia intermedia, dan talasemia mayor. Penentu daripada tingkat keparahan seseorang terhadap beta talasemia bergantung pada pola pewarisan gen yang diturunkan dari kedua orang tuanya.[4]
Pola Pewarisan beta talasemia:[4]
- Autosomal Resesif: beta talasemia diturunkan dengan cara resesif autosom, artinya seseorang harus menerima dua salinan gen HBB yang bermutasi, satu dari setiap orang tua, untuk dapat mengalami kondisi atau manifestasi yang berat dari beta talasemia, seperti yang dialami oleh individu dengan talasemia mayor atau intermedia.
- Status Pembawa (Carrier): Individu yang hanya memiliki satu salinan gen HBB yang bermutasi disebut sebagai pembawa atau carrier. Mereka umumnya tidak menunjukkan gejala atau hanya mengalami gejala ringan. Namun demikian, mereka tetap bisa mewariskan gen tersebut kepada anak-anaknya.
Pendekatan Komprehensif dalam Penanganan Beta Talasemia: Dari Transfusi Hingga Terapi Gen
Pada umumnya, seseorang yang membawa sifat beta talasemia tidak menunjukkan gejala dan tidak memerlukan pengobatan khusus. Meski begitu, konseling genetik sangat disarankan karena ada risiko mewariskan gen mutasi tersebut kepada keturunannya.[5]
Sementara itu, individu dengan beta talasemia intermedia mungkin mengalami anemia ringan, tetapi umumnya tetap bisa menjalani kehidupan yang normal dengan pemantauan medis secara berkala. Dalam beberapa kasus, transfusi darah mungkin diperlukan, dan suplemen asam folat sering kali direkomendasikan.[5]
Di sisi lain, penderita beta talasemia mayor memerlukan perhatian medis yang lebih intensif. Mereka cenderung bergantung pada transfusi darah secara rutin sebagai bagian dari perawatan jangka panjang. Selain itu, terdapat beberapa pilihan terapi yang dapat dijajaki untuk membantu mengelola kondisi ini.[5]
Berikut ini adalah beberapa prosdeur yang dapat dilakukan dalam penanganan beta talasemia:[5]
1. Transfusi Darah
Tujuan utama transfusi darah pada seseorang dengan beta talasemia terutama yang mayor adalah guna mengatasi anemia berat yang terjadi dengan menggantikan sel darah merah yang sehat. Namun, perlu diperhatikan bahwa transfusi darah rutin yang berkelanjutan dalam jangka waktu yang panjang dapat menyebabkan akumulasi zat besi berlebih dalam tubuh, yang dapat merusak organ seperti hati dan jantung jika tidak dikelola dengan baik.
2. Terapi Kelasi Besi / Iron Chelation
Terapi kelasi besi / iron chelation penting bagi pasien yang menerima transfusi darah secara rutin, karena transfusi menyebabkan kelebihan zat besi dalam tubuh. Zat besi berlebih dapat menumpuk di organ vital, yang mengarah pada komplikasi serius, seperti penyakit hati dan gagal jantung. Terapi kelasi zat besi melibatkan penggunaan obat yang dapat mengikat zat besi berlebih dan mengeluarkannya melalui urin atau tinja.
Terapi ini biasanya berlangsung seumur hidup pada pasien yang rutin melakukan transfusi, karena transfusi darah secara terus menerus dapat menyebabkan penumpukan zat besi.
3. Transplantasi Stem Cell atau Bone Marrow (Sel Punca atau Sumsum Tulang)
Terapi ini bertujuan menggantikan sel punca pembentuk darah yang bermasalah dengan sel sehat yang mampu memproduksi hemoglobin normal. Proses transplantasi sel punca atau sumsum tulang melibatkan penggantian sumsum tulang pasien—yang berfungsi memproduksi sel darah—dengan sumsum tulang sehat dari donor yang memiliki kecocokan genetik, biasanya kerabat dekat seperti saudara kandung.
Namun, terapi ini memiliki tantangan besar. Menemukan donor yang genetiknya cocok sering kali menjadi kendala. Selain itu, prosedurnya memiliki risiko serius, termasuk penyakit graft-versus-host (GVHD), di mana sel donor justru menyerang jaringan tubuh pasien, menimbulkan komplikasi yang tidak bisa diabaikan.
4. Terapi Gen

Proses terapi gen.[5]
Terapi gen kini dianggap sebagai terobosan baru dalam pengobatan beta talasemia, dengan cara langsung menargetkan akar penyebab penyakit ini. Terapi ini bekerja dengan memperbaiki gen HBB, yang berperan dalam produksi hemoglobin abnormal pada penderita. Pendekatan ini menawarkan harapan baru dalam mengatasi kondisi yang sebelumnya memerlukan pengobatan jangka panjang dan transfusi darah rutin.
Meskipun masih berada dalam tahap uji klinis, hasil awal menunjukkan potensi yang menggembirakan. Beberapa pasien dilaporkan berhasil mengurangi kebutuhan transfusi darah, bahkan ada yang tidak lagi membutuhkannya sama sekali. Namun, terapi gen ini belum tersedia secara luas dan biayanya masih tinggi. Selain itu, penelitian lebih lanjut diperlukan untuk memastikan keamanan serta efektivitasnya dalam jangka panjang.
Pentingnya Deteksi Dini Beta Talasemia
Deteksi dini beta talasemia melalui screening sangat krusial untuk mengidentifikasi individu yang membawa gen ini, mencegah munculnya kasus baru, serta meningkatkan pengelolaan dan pengobatan yang tepat. Screening ini terutama ditujukan bagi mereka yang diduga sebagai carrier, karena mereka sering kali tidak menunjukkan gejala, namun memiliki gen yang dapat diwariskan kepada keturunannya.[1, 6]
Melalui deteksi dini, kita dapat mengenali pembawa gen beta talasemia yang penting untuk konseling genetik dan perencanaan kehamilan di masa mendatang. Selain itu, identifikasi pembawa gen dapat membantu mengurangi jumlah anak yang lahir dengan beta talasemia, sehingga program screening pra-nikah dan prenatal menjadi sangat penting. Deteksi dini juga membuka peluang untuk intervensi lebih awal, seperti transfusi darah rutin bagi anak-anak dengan talasemia mayor, guna mencegah anemia berat dan komplikasi yang dapat timbul.[6]
Baca juga: Talasemia: Penyakit Kelainan Darah Bawaan & Cara Mencegahnya
Solusi Andal Deteksi Talasemia: Akurat, Cepat dan Portabel
Salah satu alat yang mendukung deteksi ini adalah Hemex Gazelle Hb Variant, sebuah perangkat diagnostik in-vitro modern yang dirancang untuk mengenali varian hemoglobin, termasuk beta talasemia, dengan cepat dan akurat. Menggunakan teknologi elektroforesis asetat selulosa dalam microchip, alat ini memberikan solusi yang andal dan terjangkau untuk pengujian di titik layanan kesehatan.

Hemex Gazelle, Solusi Andal Deteksi Talasemia.
Dengan kemudahan penggunaan dan portabilitasnya, Hemex Gazelle ideal untuk digunakan di klinik, rumah sakit, maupun lokasi terpencil. Hanya dalam delapan menit, alat ini dapat memberikan hasil analisis dengan sampel darah yang sedikit, sehingga memfasilitasi pengelolaan dan tata laksana beta talasemia yang lebih cepat dan efektif. Untuk informasi selengkapnya mengenai Hemex Gazelle, silakan dapat mengunjungi halaman berikut ini:
Referensi Artikel
- Forni, G. L., Grazzini, G., Boudreaux, J., Agostini, V., & Omert, L. (2023). Global burden and unmet needs in the treatment of transfusion-dependent beta talasemia. Frontiers in Hematology, 2, 1187681. https://doi.org/10.3389/frhem.2023.1187681.
- Wahidiyat, P. A., Sari, T. T., Rahmartani, L. D., Iskandar, S. D., Pratanata, A. M., Yapiy, I., Setianingsih, I., Atmakusuma, T. D., & Lubis, A. M. (2022). Thalassemia in Indonesia. Hemoglobin, 46(1), 39-44. https://doi.org/10.1080/03630269.2021.2023565.
- Saad, H. K. M., Taib, W. R. W., Ab Ghani, A. S., Ismail, I., Al-Rawashde, F. A., Almajali, B., Alhawamdeh, M., Abd Rahman, A. A., Al-Wajeeh, A. S., & Al-Jamal, H. A. N. (2023). HBB Gene Mutations and Their Pathological Impacts on HbE/β-Thalassaemia in Kuala Terengganu, Malaysia. Diagnostics (Basel, Switzerland), 13(7), 1247. https://doi.org/10.3390/diagnostics13071247.
- Langer AL. Beta-Thalassemia. 2000 Sep 28 [Updated 2024 Feb 8]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://ncbi.nlm.nih.gov/books/NBK1426/.
- Ali, S., Mumtaz, S., Shakir, H. A., Khan, M., Tahir, H. M., Mumtaz, S., Mughal, T. A., Hassan, A., Kazmi, S. A. R., Sadia, Irfan, M., & Khan, M. A. (2021). Current status of beta-thalassemia and its treatment strategies. Molecular genetics & genomic medicine, 9(12), e1788. https://doi.org/10.1002/mgg3.1788.
- Tuo, Y., Li, Y., Li, Y., Ma, J., Yang, X., Wu, S., Jin, J., & He, Z. (2024). Global, regional, and national burden of thalassemia, 1990-2021: a systematic analysis for the global burden of disease study 2021. EClinicalMedicine, 72, 102619. https://doi.org/10.1016/j.eclinm.2024.102619.